Background and Theory of the Nuclear Overhauser Effect
The nuclear Overhauser effect (NOE) is manifested by the transfer of nuclear spin polarization from one nuclear spin population to another via cross-relaxation. It is a commonly encountered phenomenon, observed by nuclear magnetic resonance (NMR) spectroscopy.
The theoretical basis for NOE was described1 and experimentally verified by Anderson and Freeman in 1962. NOE is derived from the seminal work of Albert Overhauser, who proposed2 in 1953 that nuclear spin polarization could be enhanced by the microwave irradiation of the conduction electrons in certain metals. The general Overhauser effect was first demonstrated3 experimentally by Carver and Slichter, also in 1953. In 1963 Kaiser reported4 an NMR experiment wherein the spin polarization was transferred from one population of nuclear spins to another, rather than from electronic spins to nuclear spins. However, the theoretical basis and the relevant Solomon equations' had already been published5 by Ionel Solomon in 1955
NOE Principles and Applications
NOE has been shown to be versatile in NMR spectroscopy for helping to elucidate6 organic chemical structures. In this context, the NOE differs from the phenomenon of spin-spin coupling in that the NOE occurs through space, not through chemical bonds. Consequently, atoms that are in close proximity to each other can exhibit NOE, whereas spin coupling is observed only when the atoms are connected by 2-3 chemical bonds. The inter-atomic distances derived from the observed NOE can often help to confirm a precise molecular conformation, i.e. the three-dimensional structure of a molecule.
In 2002, Kurt Wüthrich was awarded the Nobel Prize in Chemistry for demonstrating that NOE could be exploited using two-dimensional NMR spectroscopy to determine the three-dimensional structures of biological macromolecules in solution.7,8
Some examples of two-dimensional NMR experimental techniques exploiting NOE include:
NOESY Nuclear Overhauser Effect Spectroscopy
HOESY Heteronuclear Overhauser Effect Spectroscopy
ROESY Rotational Frame Nuclear Overhauser Effect Spectroscopy
TRNOE Transferred Nuclear Overhauser Effect Spectroscopy
DPFGSE-NOE Double Pulsed Field Gradient Spin Echo NOE experiment
Some Specific Examples of Structural Elucidation Using Nuclear Overhauser Effect Spectroscopy (NOESY)
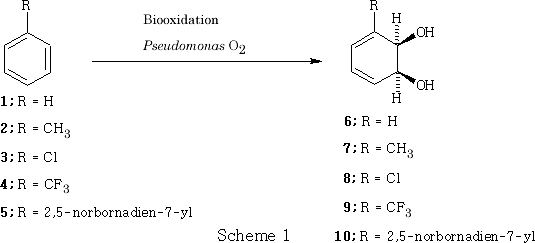
The conversion of benzene into cyclohexa-3,5-diene- cis -1,2-diol (4) by Pseudomonas spp was initially investigated (9) in pioneering work by Gibson. The successful scale up of the biotransformation by Taylor et al. (10,11) that led to the material becoming commercially available for synthetic exploitation. The bio-oxidation of simple benzene derivatives such as toluene (2) and chlorobenzene 3 can also be performed using Pseudomonas, Scheme 1 . A bonus is accrued in stereochemical terms in that these and other similar diene- cis -diols obtained ( e.g. 9 and 10 ) are optically active. In those cases, where the absolute configuration has been elucidated, the 1(S) isomer has been shown to be the major or sole enantiomer.
Early exploration of the synthetic potential of various diene- cis -diols was carried out by the research groups of Steve Ley,12 Tomas Hudlicky13 and Stanley Roberts.14 ( n.b. References 12-14 represent only a small fraction of the total work published by these authors in this field.)
This section examines the use of NOESY in the structural elucidation of some cycloadducts, derived from 1,2-isopropylidenedioxycyclohexadienes. The synthetic work is to be found in a report by the Roberts group.14 The relevant NOESY analysis is to found in the work of Williams.15 The various adducts prepared by the Roberts group were designed to be potential intermediates for the synthesis of biologically active compounds.
Synthetic Procedures Involving Cycloadditions
Cyclohexadiene- cis -diols 6, 9 and 10 were prepared by biotransformation and subsequently protected as the corresponding acetals 12-14 , Scheme 2 , utilising 2,2-dimethoxypropane 11 under acid catalysis. These protection procedures usually proceed in good yield.
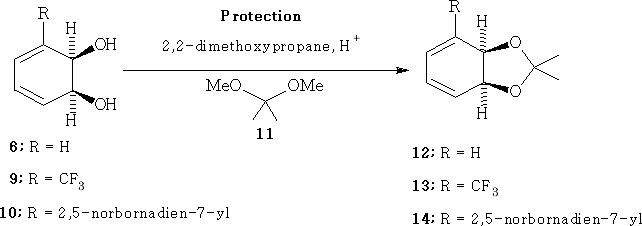
Scheme 2
Diels-Alder Cycloaddition Reactions
Nitrosobenzene 15 underwent cycloaddition in aqueous medium at ambient temperature with the 7-substituted norbornadiene 14 to give adduct 16 in 66% yield.
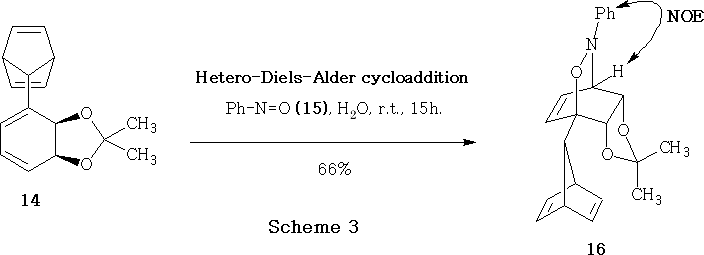
The mode of addition of the nitroso group during the hetero-Diels-Alder reaction was indicated by NOE experiments,14 which demonstrated that the phenyl ring was located close to the proton CH-N< as shown in structure 16 , Scheme 3 .
Many of the cycloadditions (with the exception of Scheme 3 ) involving various 1,2-isopropylidenedioxycyclohexadienes are described in detail, along with NOESY analysis of the products, in the PhD thesis of J. O. Williams,15 and in the PhD theses of C. A. Pittol and P. W. Sutton.
As might be expected,16 N -ethylmaleimide 17 added at ambient temperature in stirred benzene solution to the diene 12 in a non-regioselective manner to give the adducts 18 and 21 (96% combined yield) in the ratio 3 : 2.17 Similarly the trifluoromethyl diene 13 was refluxed in benzene with dienophile 17 to give two products 19 and 22 (88% combined yield) in the ratio 5 : 4, while the 2,5-norbornadien-7-yl derivative 14 underwent cycloaddition in aqueous medium at ambient temperature to give the adducts 20 and 23 (69% combined yield) in the ratio 2 : 3.
In the Diels-Alder reactions depicted in Scheme 4 , there are, in theory, four ways the dienophile 17 can add to each of the dienes 12-14 . The dienophile N -ethylmaleimide 17 can add either anti (adducts 18-20 ) or syn (adducts 21-23 ) with respect to the cis -isopropylidenedioxy group of each diene.

The dienophile N -ethylmaleimide 17 adds to give exclusively endo (adducts 18-23, Scheme 4 ) with respect to the cis -diene moiety of substrates 12-14 . These endo additions by dienophile 17 are favoured due to the extended conjugated p -electron system associated with the two amide carbonyl groups disposed cis to the dienophilic double bond of maleimide 17 . The kinetic endo effect in the Diels-Alder reaction is discussed in general terms by Gilchrist and Storr18 and Fringuelli and Taticchi.19 The preference for endo Diels-Alder addition has been rationalised by invoking stabilizing secondary orbital interactions,20-23 inductive24 or charge transfer interactions,25 and by the geometrical overlap relationship of the p -orbitals at the primary centres.26 Formation of exo/anti and exo/syn addition of dienophile 17 to dienes 12-14 is discouraged by such electronic considerations, while the formation of any endo/syn adducts is also precluded due to acute steric crowding with the acetonide group during the approach of dienophile 17 and in the hypothetical resultant product.
There is an aspect of the stereochemistry of the Diels-Alder reaction which becomes important when the two faces of the p -bond system of the interacting diene and/or dienophile are not equivalent.27-32 This phenomenon manifests itself whenever the plane through the multiple bond system of either one or both of the reactants does not represent a plane of symmetry. In this case the cycloaddition gives rise to adducts that are diastereomers. The two ways of addition are called syn and anti with respect to the group, or structural moiety, which makes the two faces different. The syn and anti additions of a generalised dienophile to a monosubstituted cyclopentadiene are illustrated in Figure 1 .
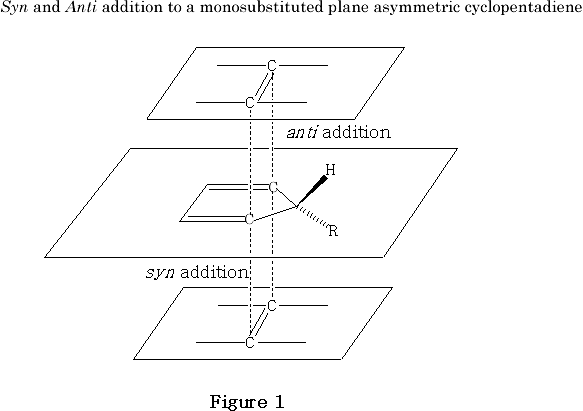
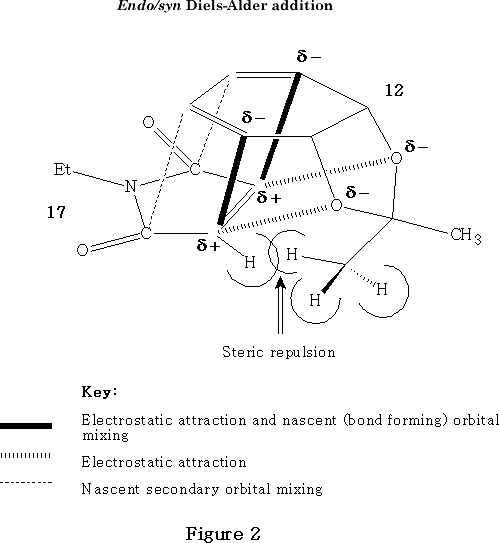
It may be surmised that many different interactions can be operating to determine syn/anti selectivity during the early stages of transition state formation in appropriate Diels-Alder reactions, depending on the nature of the atom centres involved in the cycloaddition process and relevant substituents in either the diene or dienophile. The suspected interplay of various conflicting forces and effects is shown diagrammatically in Figure 2 , which depicts the dienophile 17 lining up during the early stages of the transition state prior to endo/syn cycloaddition to diene 12 .
NOESY Analysis of Cycloadducts
NOESY Analysis of the Endo/Anti and Endo/Syn Adducts (18) and (21)
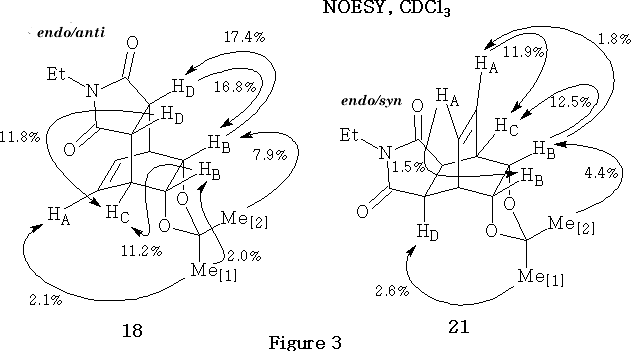
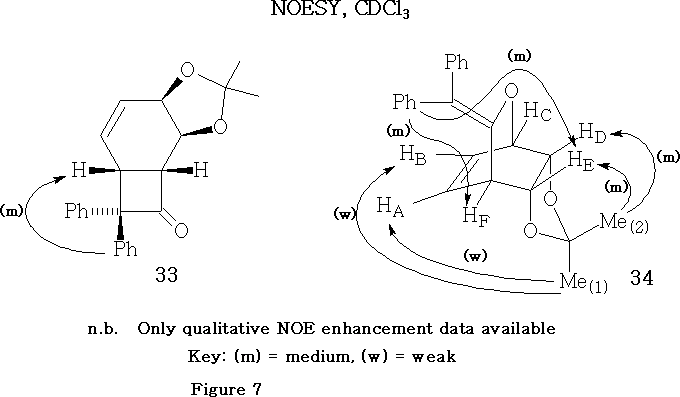
The components of the pairs of endo/anti and endo/syn products (18/21 and 19/22, Scheme 4) were distinguished,15 using NOESY in deuteriochloroform. The simpler example is the unsubstituted pair 18/21 shown in Figure 3 .
The curved arrows point from the irradiated proton(s) to the enhanced proton(s). Enhancements are given as percentages. For example, in compound (18, Figure 3) the signal associated with proton HC is enhanced by 11.8%, when proton HD is irradiated.
Key, defining NOE relationships, exhibited by the endo/anti adduct 18 , are the enhancement of methine proton HA by irradiation of the endo methyl group Me [1] and the mutual enhancements observed between protons H B and H D , when irradiated in turn.
Key, defining NOE relationships, exhibited by the endo/syn adduct 21 , are the enhancement of proton H D by irradiation of the endo methyl group Me[1] and the mutual enhancements observed between protons HA and HB , when irradiated in turn.
It may be noted that the above adducts 18 and 21 , (Scheme 4 and Figure 3) are meso compounds, because they each contain a plane of symmetry. Due to this symmetry property the magnitudes of the NOE percentage enhancements, caused by the saturation of the isopropylidene methyl groups Me[1] and Me[2] , which lie on the plane of symmetry, have been divided by a factor of two.
It must be remembered that in general NMR data, including NOESY spectra, are only relative and can only provide supportive rather than definitive evidence with respect to any proposed structure. X-ray crystallographic analysis may be considered conclusive.
NOESY analysis of the endo/syn trifluoromethyl substituted adduct (22)
The endo/syn trifluoromethyl substituted adduct 22 was obtained along with the endo/anti adduct 19 as described in (Scheme 4, R = CF3 ). Adduct 22 was analysed using NOESY (Figure 4) . Unlike the unsubstituted endo/syn adduct (21, Scheme 7) , acetonide 22 is not a meso compound. The presence of the trifluoromethyl group breaks the plane of symmetry, which exists in the analogous endo/syn adduct 21 . Consequently, the NOE enhancements associated with the methyl groups of the isopropylidene moiety in adduct 22 do not need to be adjusted.
Key, defining NOE relationships exhibited by the endo/syn trifluoromethyl substituted adduct (22, Figure 4) are the enhancement of protons HC and HD by irradiation of the endo methyl group Me[1] and the signal enhancement observed for proton HG when methine proton HA was irradiated.
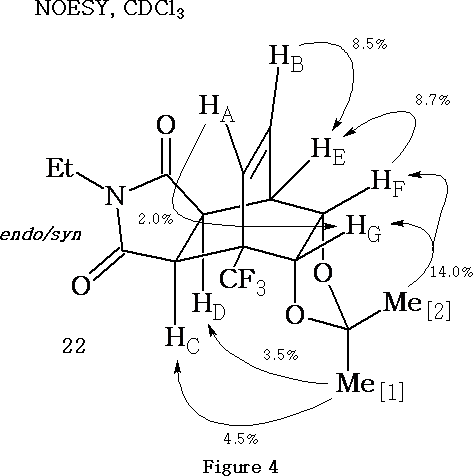
The NOESY results depicted in Figure 4 for adduct 22 are consistent with the proposed endo/syn structure. Confirmation of this structural proposal was later provided by X-ray crystallographic analysis.
[6 + 4] Cycloadditions Involving Tropone (24)
One of the triumphs of the concept of the Conservation of Orbital Symmetry, described by Woodward and Hoffmann, was the prediction33 in 1962 of the viability of a concerted [6+ 4] cycloaddition reaction. Four years later this prediction was born out34 by Cookson et al. , who combined cyclopentadiene 24 with tropone 25 to give exo adduct (26, Scheme 5) . These results were shortly afterwards confirmed35 by Itô et al. , who obtained the same product 26 , using slightly different reaction conditions.
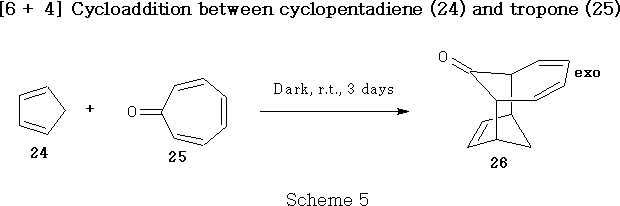
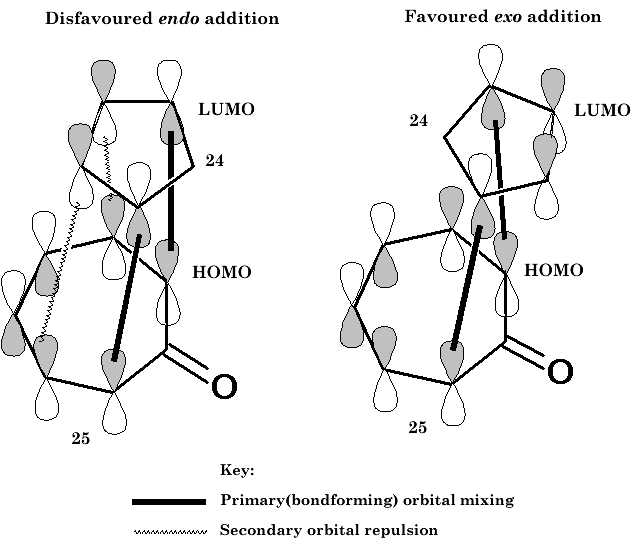
It may be noted that the stereochemistry of adduct 26 is exo , in line with secondary orbital considerations predicted33 by Woodward and Hoffmann. In some cases the secondary orbital interactions among unsaturated centres, involved in a concerted cycloaddition reaction, are such as to raise, rather than lower, the energy of the of the endo transition state and lead to a preference for exo addition, insofar as orbital symmetry factors are dominant. Inspection of the orbital diagrams in Figure 5 for the symmetry allowed [6 + 4] addition indicates that it is such a case.
Tropone 25 underwent smooth [6 + 4] cycloaddition with diene 12 , affording only the exo/anti adduct (27, Scheme 6).14,15

The exo/anti configuration of the product 27 was consistent with the NOESY analysis15 shown in Figure 6 . It may be noted that the magnitude of NOE enhancements in Figure 6 are only quoted to the nearest 0.5%.
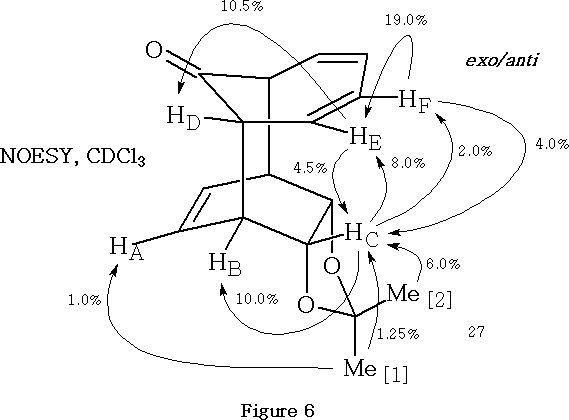
Key, defining NOE relationships, exhibited by the exo/anti adduct (27, Figure 6) , are the enhancement of methine proton HA by irradiation of the endo methyl group Me[1] , the mutual enhancements observed between methine proton HE and proton HC when irradiated in turn and the mutual enhancements observed between methine proton HF and proton HC when irradiated in turn .
The structure of adduct 27 was confirmed by X-ray crystallography.
[2 + 2] Cycloadditions Involving Diphenylketene (29)
The most synthetically useful reaction of ketenes is the [2 + 2] cycloaddition to form a four-membered ring. This reaction constitutes one of the few routes to synthetically versatile cyclobutanone rings.36
In 1969 Huisgen and Otto demonstrated the addition of diphenylketene 29 to cyclopentadiene 24 to give cyclobutanone 30.37 This process was employed in modified form by Winders38 in 1988, generating diphenylketene in situ , giving the desired adduct (29, Scheme 7) in good yield.
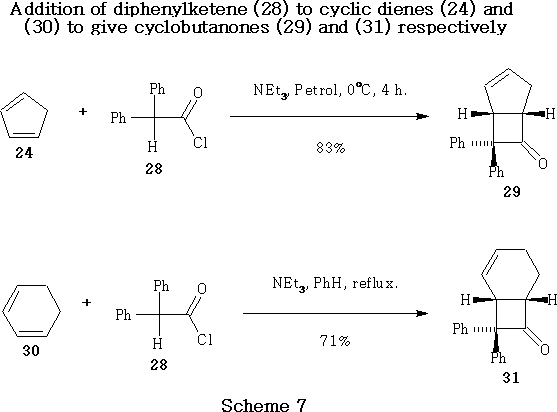
The less reactive cyclohexa-1,3-diene 31 underwent [2 + 2] cycloaddition with diphenylketene 29 , generated in situ in refluxing benzene, to give cyclobutanone (32, Scheme 7) in moderately good yield.37,38
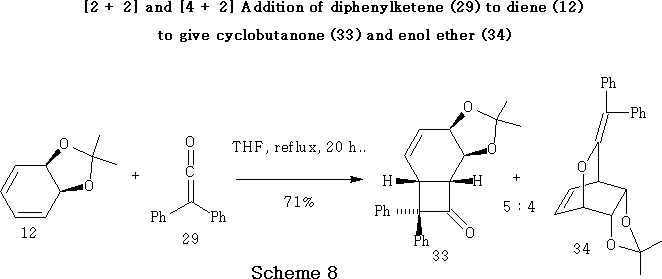
Addition of Diphenylketene (29) to Diene (12)
Diphenylketene 29 was prepared ex situ by the method39 of Taylor, McKillop and Hawks, according to the mechanism shown in Scheme 7 . Diphenylacetyl chloride 28 was treated with triethylamine in diethyl ether for one hour at room temperature to give the desired ketene 29 in 53% yield, after the precipitate of triethylamine hydrochloride had been removed by filtration under nitrogen.15
In contrast with the examples shown in Scheme 7 , diphenylketene 29 reacted with diene 12 in a non-specific manner to give not only the expected [2 + 2] adduct 33 as the major product but also a slightly lesser amount of [4 + 2] adduct 34.14,15
Ketenes generally react with conjugated dienes in a [2 + 2] fashion to produce cyclobutanones.40,41 [4 + 2] Cycloaddition across the carbon-to-carbon double bond of a ketene is rare and has only been observed with the reactions of sterically congested diene systems.42 Woodward and Hoffmann have speculated43 that the [2 + 2] pathway occurs preferentially through the initial participation of the low energy ? *C=O orbital of the ketene, followed by a concerted [ p2s + p2a ] cycloaddition of the two components. Alternatively, a concerted [ p2s ( p2s + p2s )] pathway44 and an ionic pathway have also been proposed.45
In contrast, [4 + 2] cycloadditions involving the carbon-to-oxygen double bond of a ketene are less frequently encountered. Such reactions were thought to occur only in a limited number of cases, usually involving highly strained or sterically encumbered dienes.46-48
The unsubstituted adducts 33/34 were stable in boiling THF, but readily interconverted in refluxing octane or toluene to afford an approximately equimolar mixture of the adducts 33 and 34 , fortuitously expanding the synthetic potential of the process.

The Roberts group reported 49,50 that a variety of cyclohexadiene- cis -diol acetal derivatives 12, 35-38 react with diphenylketene 29 in refluxing tetrahydrofuran (THF) to afford the [2 + 2] and [4 + 2] cycloaddition products 33, 39-42 and 34, 43-46 , respectively.
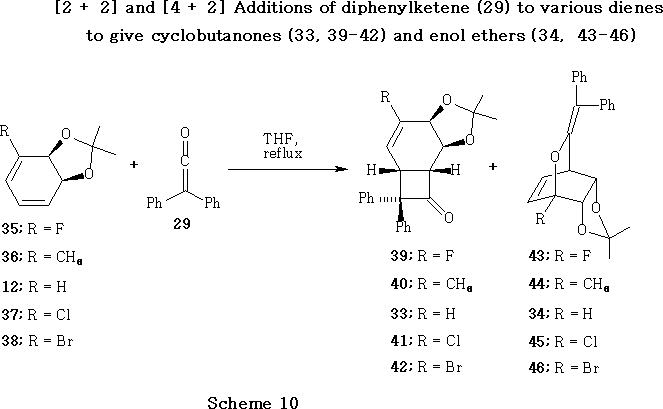
Addition of diphenylketene 29 to fluoro and methyl dienes 35 and 36 were shown49,50 to favour the formation of the [4 + 2] cycloaddition products 43 and 44 , whereas the chloro and bromo dienes 37 and 38 favoured the [2 + 2] cycloaddition products 45 and 46 ; the unsubstituted diene 12 gave almost equimolar quantities of the adducts 33 and 34 .
The methyl adducts 40 and 44 , the chloro adducts 41 and 45 and the bromo adducts 42 and 46 readily isomerise in refluxing THF or octane to afford similar product ratios to those obtained from the initial cycloadditions with diphenylketene 29 . Contrastingly, the unsubstituted adducts 33 and 34 were both stable in boiling THF, but readily interconverted in refluxing octane or toluene to regenerate an equimolar mixture of adducts 33 and 34 . Worthy of note is the fact that the fluoro[4 + 2] adduct 43 was found to be stable in both refluxing THF and octane, but the fluoro[2 + 2] adduct 39 was readily converted into the adduct 43 under reflux in these solvents.
The NOESY results15 for the fluoro[4 + 2] adduct 43 are shown in Figure 8 . Of pivotal importance for structural determination is the relationship between proton HE and protons HB , and HC and Ph[1]. It may be inferred from these relationships that the enol oxygen is bonded to the C-F carbon atom. The proposed overall structure for the Diels-Alder adduct 43 is consistent with the NOESY evidence.
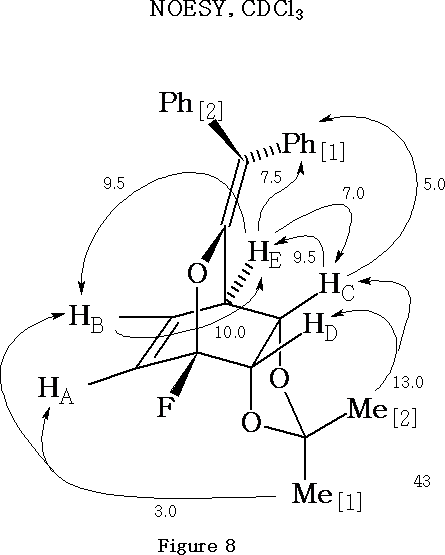
It may be noted that the magnitude of NOE enhancements in Figure 8 are only quoted to the nearest 0.5%.
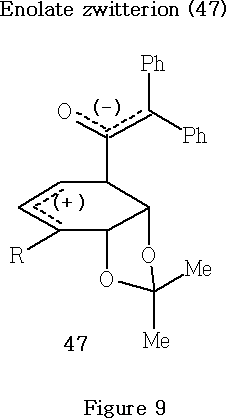
Tidwell40,41 has proposed that the reaction of cis- isopropylidene-cyclohexadienes ( e.g. 12, 35-38 ) with diphenylketene 29 , and the thermal isomerisation reactions of the resultant adducts, probably occur by an ionic mechanism involving the zwitterionic intermediate 47 . See also Reference 51 .
Addition of Methylphenylketene (48) to Diene (12)
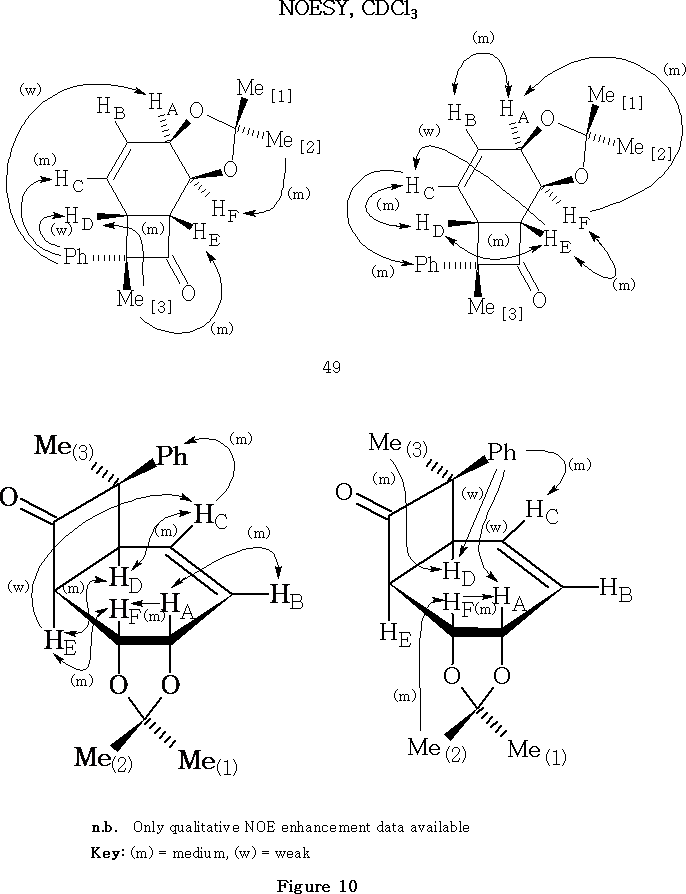
Methylphenylketene 48 was prepared39 by a modified version of the method of Taylor et al. Methylphenylketene 48 underwent [2 + 2] cycloaddition with diene 12 to give as the two main products cyclobutanones 49 and 50 in 54% and 5% overall yield respectively.15,51
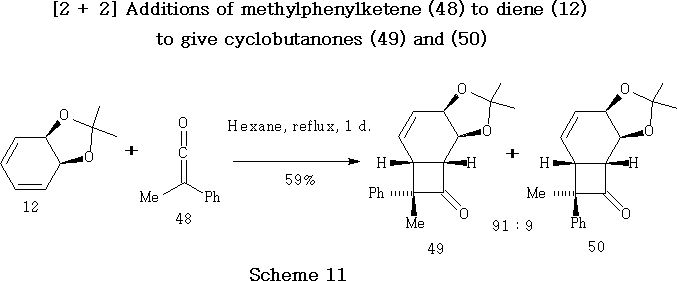
The fact that the sterically crowded endo -phenyl adduct 49 is the major product is presumed to be a consequence of what is sometimes referred to as a masochistic steric effect , thought to be consistent with suggestions of an antarafacial [ p2s + p2a ] ketene cycloaddition.52
The proposed overall structure for the [2 + 2] endo- phenyl major product 49 is consistent with the NOESY evidence, Figure 10 .